Zeroing in on Zika
Pardis Sabeti, an infectious disease expert at the Broad Institute, knows how to put together a rapid response to outbreaks. When the Ebola virus ravaged West Africa in 2014, Sabeti and her team quickly worked with scientists and health workers on the ground to understand the epidemic. They helped their African partners diagnose the first case of the deadly disease in Nigeria—a key step towards containing it in Africa’s most populous country.
Sabeti’s team is now contending with a different scourge: Zika. Thanks in part to the philanthropic support of Marc and Lynne Benioff, they’ve assembled more than 110 Zika genomes from 10 countries and territories for the largest-to-date sequencing study of the virus, which will help guide a proactive response to future outbreaks.
Though Zika has faded from headlines, it hasn’t gone away—and the consequences of the outbreak, particularly in Brazil, have been devastating. The virus has been linked to microcephaly and other serious fetal brain defects; the World Health Organization reported that as of June 15, 2017, 2,722 Brazilian babies had Zika-related birth defects. Zika has also been connected to Guillain-Barré syndrome, which affects the nervous system and can cause temporary paralysis.
Although Zika had been known in Africa and Asia for decades—it was first identified near the Zika River in Uganda in 1947—the outbreak in the Americas caught health officials off guard. For a year after the virus was first detected in Brazil in 2015, there was very little genomic information available—even though it was essential to understanding and grappling with the virus. Sabeti’s group had shown with Ebola how valuable it could be to obtain and analyze genomes in the midst of an epidemic. “By having genomes during an outbreak, you can watch how the virus is changing and, in the process, make more sensitive and specific diagnostics,” said Hayden Metsky, a Ph.D. student in the Sabeti lab.
The first hurdle was obtaining genomes. Brazil’s laws bar medical samples leaving the country for research, and Sabeti’s personal connections mostly existed in Africa. Her lab overcame this by working with MIT’s Irene Bosch, a dengue researcher with a network of collaborators in the region, and reaching out to scientists in Brazil, elsewhere in Central and South America, in the Caribbean, and in Florida. Indeed, global teamwork epitomizes Sabeti’s MO wherever outbreaks occur. “Pardis has something not patentable—relationships with scientists in a lot of countries,” said Raffaella Squilloni, a Harvard Business School Blavatnik Fellow in Life Science Entrepreneurship working with Sabeti’s group.
But the group soon faced another hurdle. This time, it was technical: Zika is present in the body at much lower quantities than Ebola and many other viruses are, which makes it difficult to sequence. To tackle this challenge, Sabeti’s team tried three different techniques to analyze the viral genome sequence from human samples and squashed mosquitos.
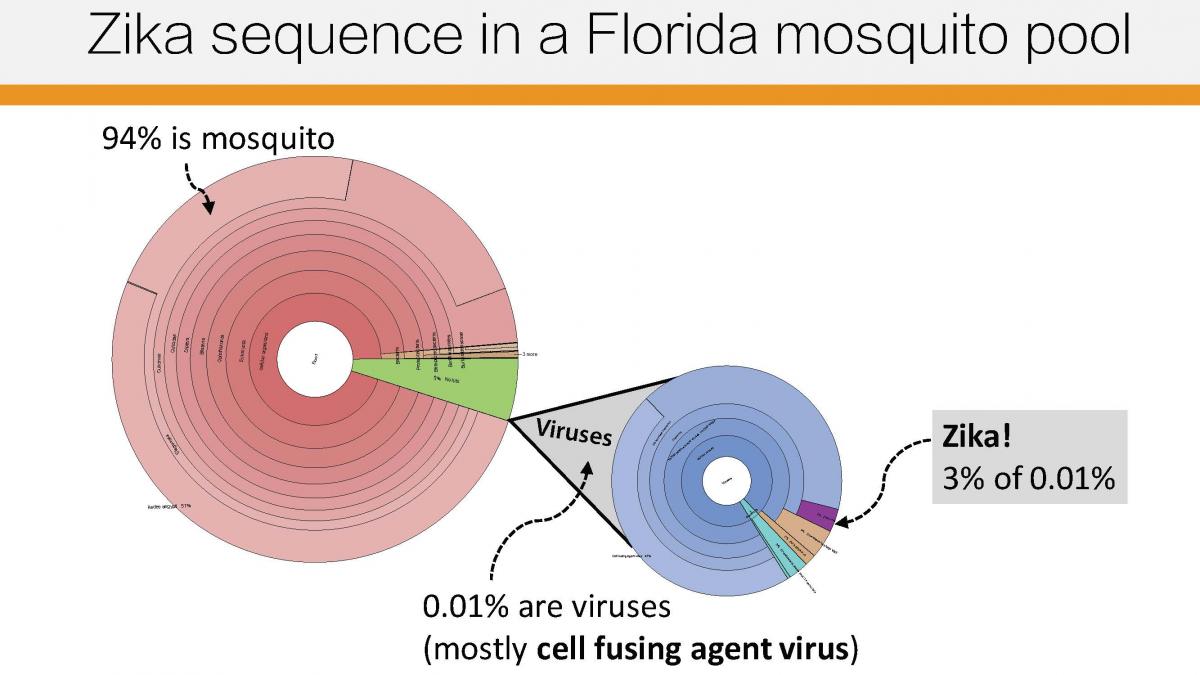 (image credit Hayden Metsky)
(image credit Hayden Metsky)
First, Metsky and fellow lab member Chris Matranga used a technique that they and their lab mates designed, which amplifies all of the genetic material in a sample, then fishes out Zika and chikungunya sequences. This approach is particularly useful for analyzing how Zika is changing inside of patients because it can detect Zika variation within samples. Plus, another team member, Mary Lynn Baniecki, was able to use the method to design Zika-specific probes as the basis for a new diagnostic for the virus.
This approach yielded 37 genomes from 66 samples. To collect even more genomes, the team supplemented their technique with a more traditional one that picks up and amplifies only Zika sequences. This more sensitive approach contributed the bulk of the 110 genomes.
Finally, the group sequenced all the viral content, in an unbiased way, in 38 samples: 30 from humans and eight from mosquitos. That allowed them to identify other viruses that were co-infecting Zika patients and mosquitos—including some that are new to science.
Together, these methods provided the single largest and most geographically diverse data set of Zika genomes available for study. Recently published in Nature, these genomes provided key insights into the outbreak: how and when Zika might have spread from country to country, and how the virus is changing over time.
Critically, the genomes revealed that Zika was lurking in Central and South America and the Caribbean for many months before it popped up on the radar. That suggests that, in the future, improved monitoring might make it possible to catch viral outbreaks early—perhaps even before viruses become outbreaks—and stop them in their tracks. In that vein, the group is developing a catch-all method to identify any virus in a sample—without necessarily knowing what you’re looking for. This pan-virus diagnostic will capture any of the approximately 300 viruses known to infect people.
The group also collaborated with other teams at the Broad to create an entirely new viral detection system for Zika and other diseases based on the genome-editing tool CRISPR. The method, known as SHERLOCK, was also partially supported by the Benioffs’ generous gift.
It’s all part of Sabeti’s vision to revolutionize the way we monitor and respond to outbreaks. “If we’ve learned anything from recent outbreaks, it’s that we need to be better prepared for the next one,” she said. “It is our hope that our work can help lay the foundation for national and global diagnostic, surveillance, and response systems that identify potential outbreaks and contain them before they spread.”
 The Sabeti Lab Zika team
The Sabeti Lab Zika team